Workflow Genomica Comparativa
Bioinformática. Generación de un árbol filogenético.
Instructor: MC José Roberto Aguirre. Colaborador y edición: Rogelio Prieto

Este documento muestra un flujo de trabajo para realizar:
a) ensamblaje de novo de genomas bacterianos
b) evaluación de calidad de los ensamblajes y
c) generación de árboles filogenéticos.
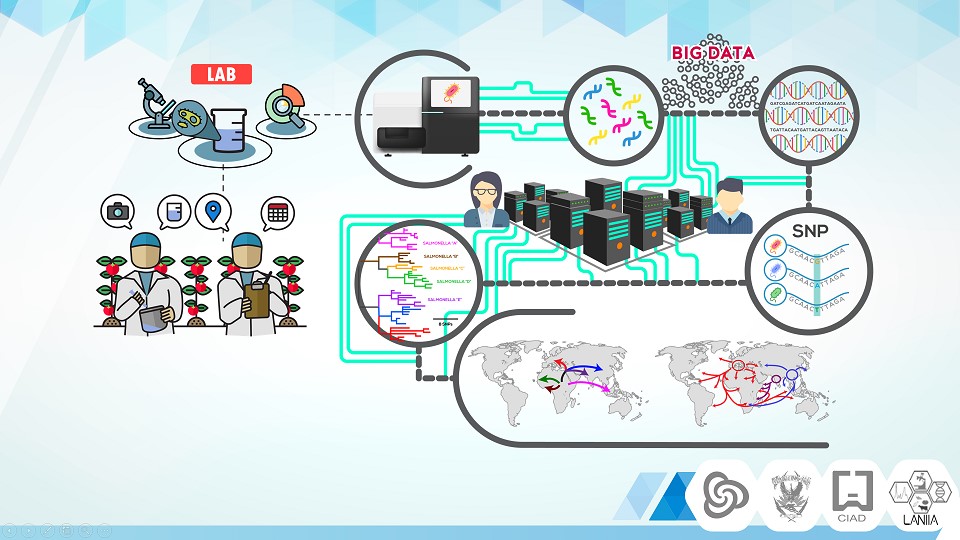
Diagrama general.
Software requerido.
| Software | Usado para | URL |
|---|---|---|
Terminal y comando wget. |
Descarga de cepas. | No aplica |
fastq-dump de NCBI SRA Toolkit. |
Conversión de formato. | https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software#header-global |
A5-miseq |
Realizar los ensambles. | https://pubmed.ncbi.nlm.nih.gov/25338718/ https://sourceforge.net/projects/ngopt/ |
A5-miseq y Spades |
Realizar los ensambles. | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3342519/ https://cab.spbu.ru/software/spades/ |
quast. |
Evaluación de la calidad de los ensambles. | https://pubmed.ncbi.nlm.nih.gov/23422339/ https://sourceforge.net/projects/quast/ |
parsnp de harvest tools |
Generar el árbol filogenético. | https://harvest.readthedocs.io/en/latest/content/parsnp.html |
iTOL: Interactive Tree Of Life. |
Visualización y edición del árbol filogenético. | https://itol.embl.de/ |
Creación de directorio.
cd ~/rprieto/reads/
mkdir genomas
Adquisición de datos.
Se descargarán genomas de cepas de Salmonella enterica encontradas en la base de datos pública del National Center for Biotechnology Information (NCBI), las cuales pertenecen a algunos de los cinco serotipos predominantes en la región.
wget ftp://ftp-trace.ncbi.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/SRR132/SRR13246501/SRR13246501.sra
Esta instrucción descarga los genomas de las cepas en formato SRA (Sequence Read Archive).
¿Cómo se obtuvo esta dirección de descarga?
La descarga de cada cepa se empieza determinando la localización siguiendo con la siguiente instrucción:
wget/FTP root: ftp://ftp-trace.ncbi.nih.gov/sra/sra-instant/reads/ByRun/sra/<SRR|ERR|DRR>/<6-caracteres-código>/<código>/<código.sra>
Donde:
<SRR|ERR|DRR>: Se refiere a la corrida de secuenciación la cual contiene todos los datos experimentales. Se utilizan los caracteresSRR|ERR|DRRpara identificar de donde procede la secuencia.SRRindica que se encuentra en NCBI,ERRpara la base de datos europea yDRRpara la base de datos japonesa. Estas tres bases comparten las secuencias, es por ello que se pueden encontrar en cualquiera de las tres bases de datos, pero con su identificador correspondiente.
Ejemplo. Para realizar la descarga del genoma SRR304976, se requiere la siguiente instrucción:
ftp://ftp-trace.ncbi.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/SRR304/SRR304976/SRR304976.sra
Donde:
wget: Es un comando del sistema operativo Linux que nos permite la descarga de archivos desde la Web.ftp://ftp-trace.ncbi.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/: Es la dirección de descarga del NCBI.SRR304: Son los primeros seis caracteres del código del genoma asignado por NCBI.SRR304976: Es el código del genoma completo asignado por NCBI.SRR304976.sra: Es el código del genoma completo asignado por NCBI sumado a la extensión.sra.
Conversión de formato: de SRA a FASTQ.
Para realizar el proceso de ensamble, se requiere convertir de formato SRA a formato FASTQ, ya que este es un formato común para compartir secuenciación de datos leídos combinando ambos tanto secuencia como puntuación de calidad asociada por base. Esta conversión se realizó utilizando la herramienta SRA Toolkit siguiendo:
$ fastq-dump –split-files <nombre del archivo SRA> -O <ruta de salida>
Donde:
fastq-dump: Es la opción para convertir información SRA en formato FASTQ.
-split-files: Separa cada archivo leído en R1 y R2. Los archivos recibirán el sufijo correspondiente al número de archivo leído.
-O: Especifica la ruta de salida, tomando de inició la ruta del directorio activo.
Ejecutar:
$ fastq-dump –split-files SRR304976.sra -O ~/rprieto/reads/genomas
Ensamble
Se realizarán ensambles utilizando los programas A5-miseq y Spades.
Ensamble con A5.
Se realizará el ensamble utilizando la herramienta A5-miseq. Se ejecutará:
a5_pipeline.pl –end=1-5 <Read 1 FastQ> <Read 2 FastQ> <out_base>
Donde:
-a5_pipeline.pl: Nombre del archivo con la instrucción del pipeline.
-end=1-5: Especifica el rango de pasos a seguir, los cuales son:
- Limpieza de los reads : Adaptadores de secuencias y regiones de baja calidad son removidos con Trimmomatic.
- Ensamble de contigs : Reads pareados y no pareados son usados para el ensamblado con el algoritmo IDBA-UD.
- Ensamble de Scaffolds : Los contigs son ensamblados para formar segmentos continuos de mayor longitud denominados Scaffolds.
- Correcciones de ensambles : Se detectan los ensambles que no corres- ponden con la distancia especificada por el proceso de mapeo.
- Re-Scaffolding final : Se realiza una ronda final para ensamblar Scaffolds que pudieron quedar desalineados.
-
out_base: Es el nombre base, es decir, el nombre en común que se le asigna a todos los tipos de archivos de salida.
| Archivo | Descripción |
|---|---|
assembly.out.ec.fastq.gz |
Reads corregidos |
assembly.out.contigs.fasta |
Contigs |
assembly.out.crude.scaffolds.fasta |
Scaffolds crudos: No han sido analizados los emsambles generados |
assembly.out.broken.scaffolds.fasta |
Scaffolds rotos: Se analizaron los ensambles pero aun no se corrigen |
assembly.out.final.scaffolds.fasta |
Scaffolds finales: Analizados y corregidos |
assembly.out.final.scaffolds.fastq |
Scaffolds finales en formato FASTQ |
assembly.out.final.scaffolds.qvl |
Valores de calidad para scaffolds finales |
assembly.out.final.scaffolds.agp |
Formato AGP que describe los scaffolds para someterlos a NCBI |
assembly.out.assembly_stats.csv |
Estadísticas de calidad separadas por tab |
Ejecutar:
a5_pipeline.pl –end=1-5 SRR13246501_1.fastq SRR13246501_2.fastq salm_1
Ensamble con Spades.
$ spades.py -k 21,33,55,77 --careful -1 SRR13246501_1.fastq -2 SRR13246501_2.fastq -o spades_output
Evaluación de la calidad de los ensambles.
El control de calidad de los ensambles sirve como una evaluación para identificar y excluir datos con problemas serios de calidad, lo que permite ahorrar tiempo en análisis posteriores. Existen algunos indicadores métricos que permiten evaluar la calidad del ensamble cuantitativamente; entre estos indicadores se encuentran:
- el número de contigs
- el número de scaffolds
- el tamaño del genoma
- el valor de N50 y
- el contenido de G+C (guaninas y citosinas)
El valor de N50 corresponde al menor de los mayores contigs que cubren la mitad del genoma y constituye un indicador acerca de la contigüidad de nuestros genomas. Un valor de N50 pequeño está asociado a un mayor número de contigs y scaffolds, lo que también se podría ver reflejado en un aumento en pares de bases en el ensamble.
Se utilizará QUAST para evaluar la calidad de los dos ensambles. Se ejecuta el programa, se le envía el ensamble generado por A5-miseq y Spades:
$ quast.py ensamble_A5.fasta ensamble_spades.fasta
Descargar otros ensambles.
Se descargarán genomas similares con la finalidad de ampliar el espacio muestral del organismo de interés. Esto permitirá contextualizar los datos analizados.
En este caso se propone la descarga de 8 genomas en formato fasta.
Ejecutar:
wget https://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/016/495/825/GCA_016495825.1_PDT000927971.1/GCA_016495825.1_PDT000927971.1_genomic.fna.gz
wget https://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/016/495/145/GCA_016495145.1_PDT000927984.1/GCA_016495145.1_PDT000927984.1_genomic.fna.gz
wget https://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/016/495/625/GCA_016495625.1_PDT000927974.1/GCA_016495625.1_PDT000927974.1_genomic.fna.gz
wget https://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/016/495/465/GCA_016495465.1_PDT000808939.3/GCA_016495465.1_PDT000808939.3_genomic.fna.gz
wget https://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/016/495/765/GCA_016495765.1_PDT000927967.1/GCA_016495765.1_PDT000927967.1_genomic.fna.gz
wget https://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/016/495/885/GCA_016495885.1_PDT000927970.1/GCA_016495885.1_PDT000927970.1_genomic.fna.gz
wget https://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/016/495/365/GCA_016495365.1_PDT000927985.1/GCA_016495365.1_PDT000927985.1_genomic.fna.gz
wget https://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/016/495/965/GCA_016495965.1_PDT000927961.1/GCA_016495965.1_PDT000927961.1_genomic.fna.gz
En necesario descomprimir cada uno de los archivos descargados.
Ejecutar:
gunzip GCA_016495825.1_PDT000927971.1_genomic.fna.gz
gunzip *.fna.gz
Generar el árbol filogenético.
Se utilizará la herramienta Parsnp que es parte del software harvest.
parsnp -r ! -d directorio/fastas -c
Donde:
-des el directorio donde se enecuentran los archivos fastas.-res la referencia, en este caso utilizamos!para indicar que tome una referencia al azar.
Visualización y edición del árbol filogenético.
iTOL(Interactive Tree Of Life) es una plataforma web que nos permitirá la visualización y edición del árbol genético. Mediante esta plataforma es posible renombrar taxas, agregar o mostrar metadatos.
https://itol.embl.de/